Fylogenetica - Theorie en Doe-Het-Zelf
Tekst en figuren: Melchior de Bruin
Foto's: Bart Jansen & Mark Ros

Fylogenetica (Phylogenetics)
Het is de laatste tijd erg druk in de wereld van de cichliden-naamgeving. Soorten verdwijnen omdat ze tóch hetzelfde blijken te zijn als een zustersoort. Andere soorten krijgen toch een eigen genus, enzovoorts. De reden voor deze onrust is een vrij nieuwe methode om de verwantschap tussen cichliden te bekijken: het maken van fylogenetische stambomen. Het woord fylogenetica is een samenvoeging van fylogenie(“afstamming van organismen”) en genetica. Het is namelijk mogelijk om stambomen te maken op basis van genetische informatie (DNA). Dit is theoretisch veel nauwkeuriger dan het maken van een stamboom op basis van verschillen in bouw of uiterlijk.
De achtergrond
Onze genetische informatie, het DNA, is grof gezien niets meer dan een hele lange reeks van vier verschillende grote moleculen. Deze moleculen heten adenine, guanine, thymine en cytosine. Afgekort: a, g, t en c. (Voor meer informatie over DNA: http://nl.wikipedia.org/wiki/DNA ). Drie DNA-moleculen coderen voor één aminozuur (eiwit-bouwsteentje).
Het DNA van cichliden is voor het overgrote deel hetzelfde. Er zijn namelijk veel biologische processen die hetzelfde zijn in elk levend organisme (denk maar aan het energie halen uit voedsel). Voor deze processen gebruiken de meeste dieren dezelfde eiwitten en enzymen. Het stukje DNA wat voor deze enzymen en eiwitten codeert is dus ook hetzelfde in bijna alle dieren.
Nou ja, eigenlijk zijn ze voor 99% gelijk… er zitten namelijk kleine verschilletjes in de DNA-reeks. Vaak zo klein dat het uiteindelijke enzym of eiwit er geen last van heeft. Er zijn bijvoorbeeld meerdere combinaties van DNA-moleculen mogelijk voor één bepaald aminozuur. Dan is het DNA wel iets anders, maar het enzym of eiwit blijft exact hetzelfde.
Fylogenie met DNA
Slimme biologen hebben enkele tientallen jaren geleden uitgevonden dat deze kleine veranderingen (mutaties) in DNA maar héél weinig voorkomen maar wel al plaatsvinden sinds het begin der leven. Deze kleine (onschadelijke) mutaties kunnen dan dus doorgegeven worden aan de soorten die later ontstaan. Gebruikmakend van (onder andere) deze theorie kun je dus het vormen van nieuwe soorten koppelen aan kleine verschillen in DNA-reeksen.
Dit plaatje geeft het principe schematisch weer:
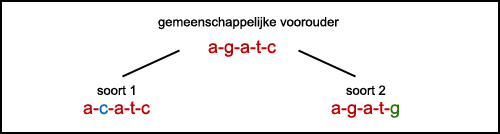
De gemeenschappelijke voorouder heeft een stukje DNA met de code “agatc”. Na duizenden jaren splitst de soort zich in twee soorten. Maar het stukje DNA is dan inmiddels een klein beetje veranderd. Zouden wij nu soort 1 en 2 opvissen en dat stukje DNA bekijken kunnen we een paar conclusies trekken.
Zo zien we dat het DNA van alle twee de opgeviste soorten op elkaar lijkt. (Ze verschillen maar 2 DNA-moleculen). De kans dat ze ooit uit één gemeenschappelijke voorouder zijn gekomen is dus heel groot. En omdat wij onze stamboom al hebben getekend, zien we dat dat inderdaad zo is. Zou de evolutie van soort 1 nog verder gaan, blijft die ene mutatie van g naar c in het DNA zitten en kunnen we die 200 soorten en 5 miljoen jaar later nog terugvinden en verbinden aan deze ene splitsing van een gemeenschappelijke voorouder in twee nieuwe soorten.
Vooral DNA uit de mitochondriën , mtDNA, is populair voor fylogenetica. De populariteit van dit type DNA is te danken aan het feit dat het alleen via de moeder overerft en daarom is de geschiedenis van dit DNA nauwkeuriger te achterhalen. Bovendien vinden er relatief veel mutaties plaats in dit type DNA.
Het maken van de stamboom
Door de DNA-reeksen te vergelijken met redelijk ingewikkelde wiskundige formules en matrixen die rekening houden met de waarschijnlijkheid van (verschillende soorten) mutaties kun je dan een mooie stamboom maken.
Een vertakking in de stamboom vind plaats wanneer de DNA-reeks volgens de berekeningen genoeg verschilt met de ander. De lengte van een tak geeft soms ook aan wanneer de splitsing heeft plaatsgevonden. De stambomen zijn iets betrouwbaarder als we aminozuren vergelijken in plaats van DNA. Maar omdat cichliden vaak zo weinig van elkaar verschillen is dat niet echt mogelijk. En zitten we dus vast aan de DNA-reeksen.
Betrouwbaarheid
Bij deze vertakkingen in de stamboom zet men vaak ook een “bootstrap”-waarde neer. Deze waarde, tussen de 1 en 100, geeft aan hoe betrouwbaar de vertakking op die plek is. Staat er bijvoorbeeld 72, wil dat zeggen dat de stamboom bij een herberekening 72 van de 100 keer op die manier uitkwam. Een bootstrap-waarde van 100 is dus het meest betrouwbaar. Kom je dus ergens een fylogenetische stamboom tegen met zulk soort waarden bij de vertakkingen, kun je zelf nagaan hoe nauwkeurig zo’n stamboom is.
Ten slotte
De fylogenetische stambomen zijn theoretisch veel betrouwbaarder dan stambomen op basis van verschillen in morfologie (uiterlijk). DNA bevat simpelweg véél meer data om te vergelijken. Maar vergeet niet dat ook fylogenetische stambomen maar hypothesen zijn. Ze kunnen dus goed zijn, fout zijn of iets er tussenin. In de praktijk is nog wel duidelijk zichtbaar dat de fylogenetica in de kinderschoenen staat. Soms kun je door een iets andere (maar goede) berekening of matrix toe te passen andere stambomen krijgen. Tot de wiskunde achter de techniek is geperfectioneerd is het dus verstandig om de bevindingen van de fylogenetici met een klein korreltje zout te nemen. Maar dat neemt niet weg dat de fylogenetica steeds meer de huidige indeling van cichliden ter discussie zal stellen.
Hoe maak ik zelf een fylogenetische boom?
Om te beginnen hebben we data nodig. Veel wetenschappers geven hun data na publicatie vrij zodat iedereen het kan gebruiken. Wij kunnen dat dus ook! Ga naar de website pubmed ( http://www.ncbi.nlm.nih.gov/sites/entrez ) en selecteer bij Search “CoreNucleotide” (of gewoon “Nucleotide”) en zoek naar een cichlide (let op: het aanbod is nog vrij beperkt). Je zoekt dan naar het DNA van een bepaald eiwit van de vissoort (in ons voorbeeld nemen we het eiwit: “NADH dehydrogenase subunit 2 (ND2) gene”). in Het is belangrijk zoekresultaten te selecteren met hetzelfde eiwit, anders kun je niet vergelijken.
Hier onder staan de zoekresultaten die wij gaan gebruiken om onze eigen fylogenetische stamboom te maken. In dit voorbeeld wil ik uitzoeken hoe het zit met verwantschap tussen vier (Neo)Lamprologus-soorten. Dit zijn de pubmed-links:
Neolamprologus multifasciatus
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nuccore&id=150406570
Neolamprologus brevis
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nuccore&id=150406566
Neolamprologus leleupi
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nuccore&id=150406518
Neolamprologus cylindricus
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nuccore&id=150406399
Oreochromis tanganicae
http://www.ncbi.nlm.nih.gov/entrez/viewer.fcgi?db=nuccore&id=20377110
En ik heb ook bewust gekozen voor vier soorten van hetzelfde geslacht, en één totaal andere soort. Deze totaal andere soort is de ‘outgroup’ die we later min of meer nodig hebben ter controle. Van de O. tanganicae weten we immers dat hij zeker niet heel verwant is aan Neolamprologus-soorten.
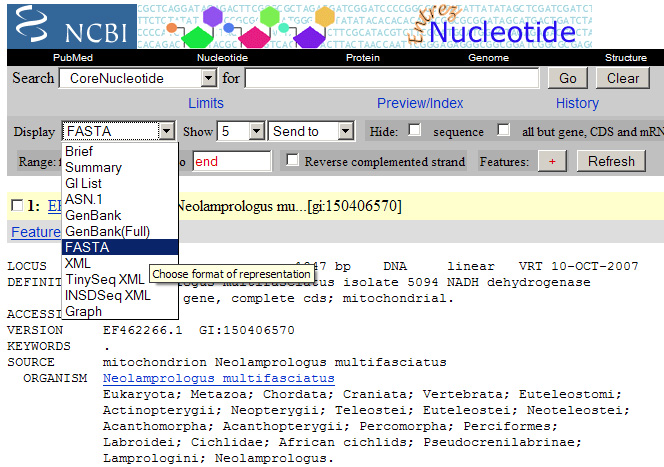
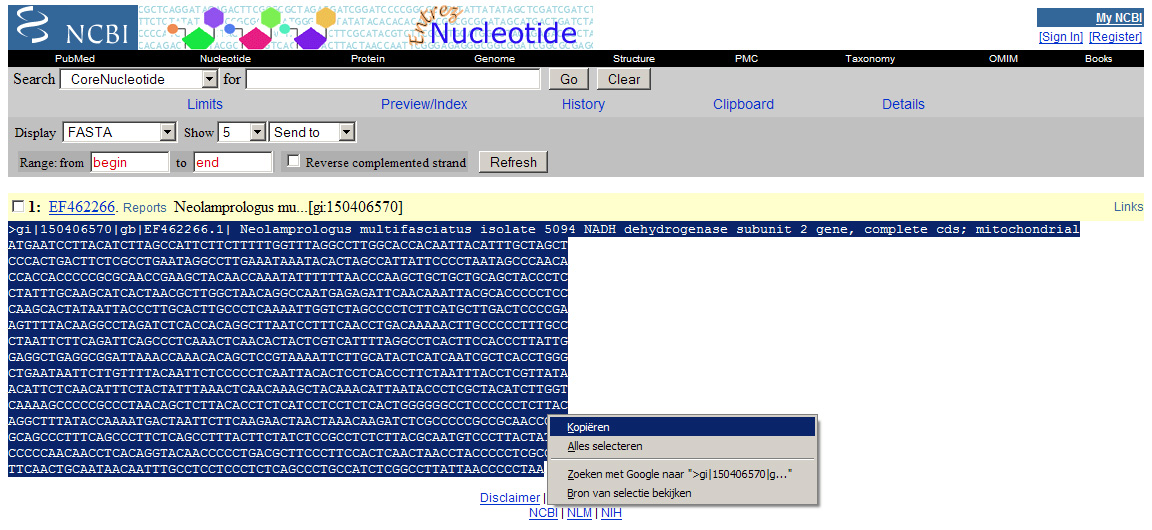
De zoekresultaten (dus de bovenstaande links) moeten we nu omzetten naar een andere vorm zodat het computerprogramma ze kan inlezen. Selecteer hiervoor bij “Display” FASTA (zie ook afbeelding 1 ). Je ziet dat alle overbodige informatie dan verdwijnt. Selecteer de informatie onder de gele balk per zoekresultaat en kopieer het (zie afbeelding 2 ) naar een notepad(kladblok)-document en zet het daar onder elkaar. (Als je deze tutorial precies volgt ziet het er uiteindelijk zo uit: fylogenetische_stamboom.txt
Nu we het voorbereidende werk hebben gedaan, kunnen we de fylogenetische stamboom echt gaan maken. Surf hiervoor naar ClustalW .
Selecteer nu alles uit je notepad(kladblok)-bestand en plak dit in het grote witte veld. Alle instellingen laten we ongewijzigd (de standaard-instellingen zijn redelijk betrouwbaar).
Klik dan op de rode “Run”-knop onderaan de pagina.
Na een paar seconden verschijnt het resultaat. Scroll naar beneden en daar staat onze eigen fylogenetische boom al! Maar omdat we graag willen zien hoe snel een bepaalde soort is afgesplitst (en dus hoe ver de ene soort van de andere afstaat) klikken we nog even op de knop: “Show as Phylogram Tree”. Dit is dan het resultaat:
 fylogenetische_stamboom.txt).
fylogenetische_stamboom.txt).
Achter deze “rare dingen” staat namelijk gewoon de Latijnse naam van de vis. (Zie ook afbeelding 4 )
Zo kom ik uiteindelijk uit op deze stamboom:

Wat deze boom wil zeggen is nu heel duidelijk te zien. Omdat ze aan dezelfde tak zitten kunnen nu concluderen dat de N. multifasciatus meer verwant is aan N. brevis dan aan z’n soortgenoten N. leleupi en N. cylindricus. (En omgekeerd, natuurlijk).
De Oreochromis tanganicae is, zoals we al hadden verwacht, een buitenbeentje. Dit geeft aan dat we onze fylogenetische boom in ieder geval grof gezien goed hebben gemaakt. Stel dat de Oreochromis plotseling volgens de stamboom nauw verwant zou zijn aan een N. multifasciatus. Dan is er iets verkeerd gegaan...
De resultaten laten mooi zien waarom we de multifasciatus en brevis liever aanduiden met “Lamprologus” in plaats van Neolamprologus. Het is nog steeds de vraag of ze wel in hetzelfde genus thuishoren.
Tot zo ver dit Doe-Het-Zelf artikel.

{kind=link}
{kind=link}
{kind=link}